[1] 12158 44[1] 44 8January 14, 2025
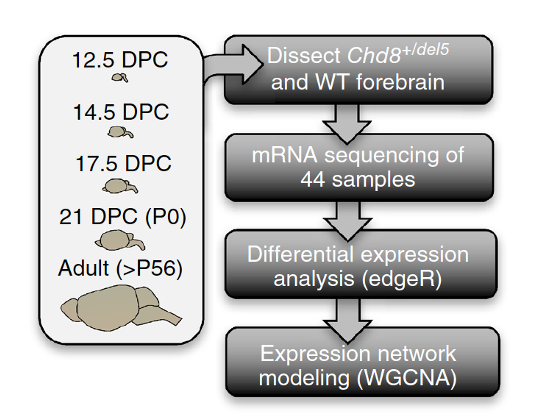
Gompers et al. (Nature Neuroscience 2017) performed RNA-seq on mice from two different genotypes (18 WT vs 26 CHD8 mutant) and 5 developmental stages
“Using a statistical model that accounted for sex, developmental stage and sequencing batch, we tested for differential expression across 11,936 genes that were robustly expressed”
We’ll use this dataset throughout this lecture to illustrate EDA
Option 1 - “Separated”: Keep main data and metadata tables separate
Pros:
Minimal startup effort / extra code
Can be compatible with downstream analysis methods (e.g. Bioconductor)
Cons:
Risky: easy to make a mistake when subsetting and/or reordering samples - extra sanity checks required
Not a convenient format for visualization since main data is separated from its metadata
Overall: not recommended
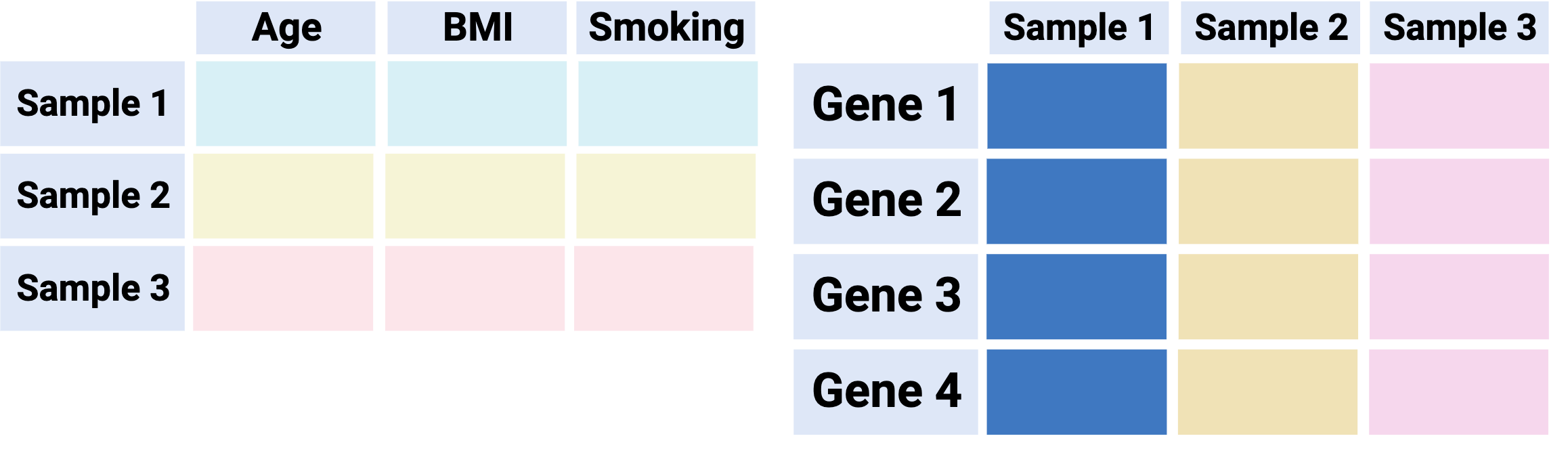
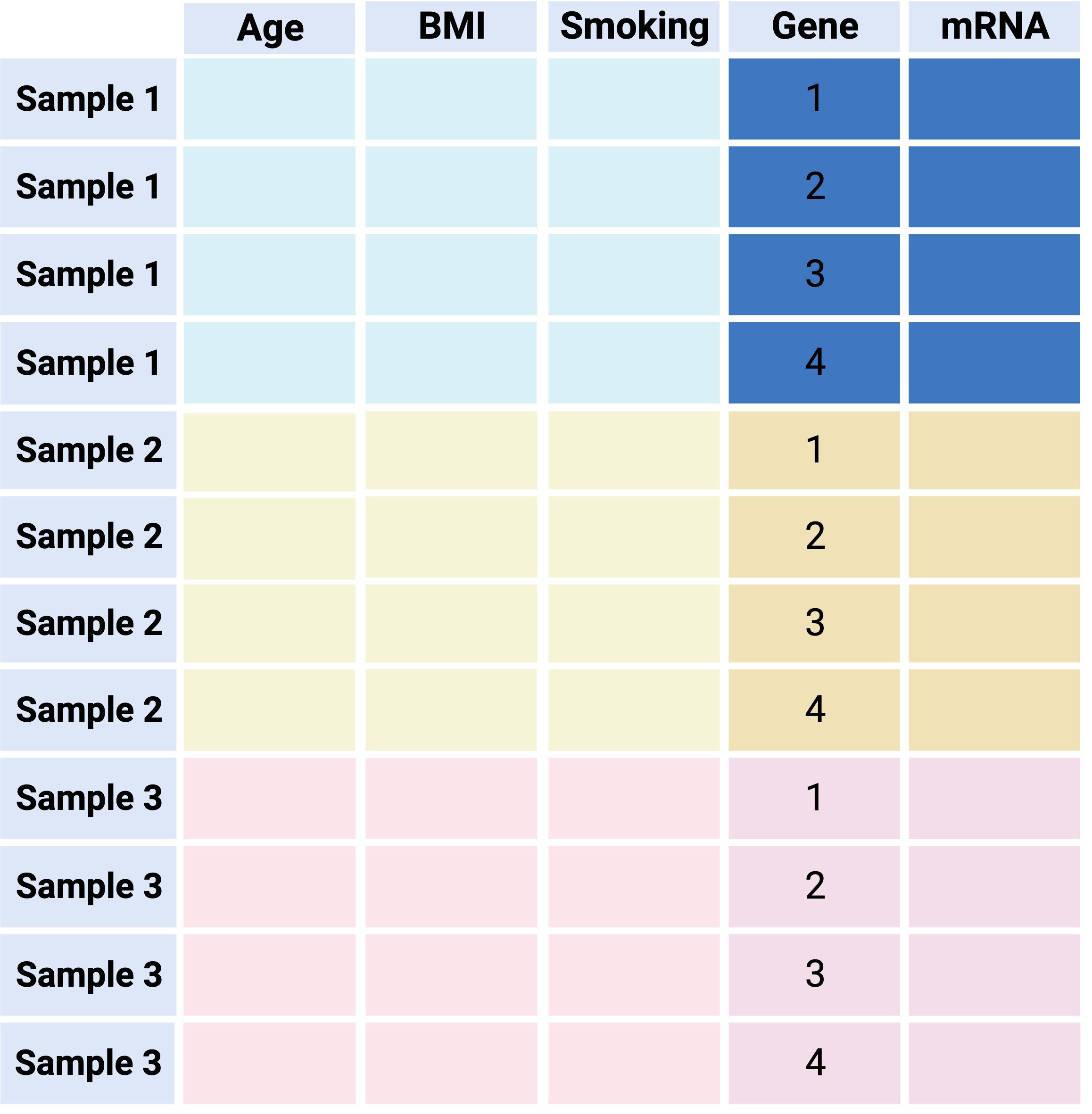
Option 2 - “The tidy way”: Combine main data & metadata into one ‘long’ table
Pros:
Unambiguous - keeps all data in one object with one row per observation (e.g. each sample/gene combination is one row, along with all its metadata)
Plays nice with tidyverse tools (e.g. dplyr manipulations, ggplot2 visualization)
Cons:
‘long’ format is inefficient data storage - sample information is repeated
Not compatible with many tools for downstream analysis (e.g. Bioconductor)
Overall: recommended for EDA/visualization
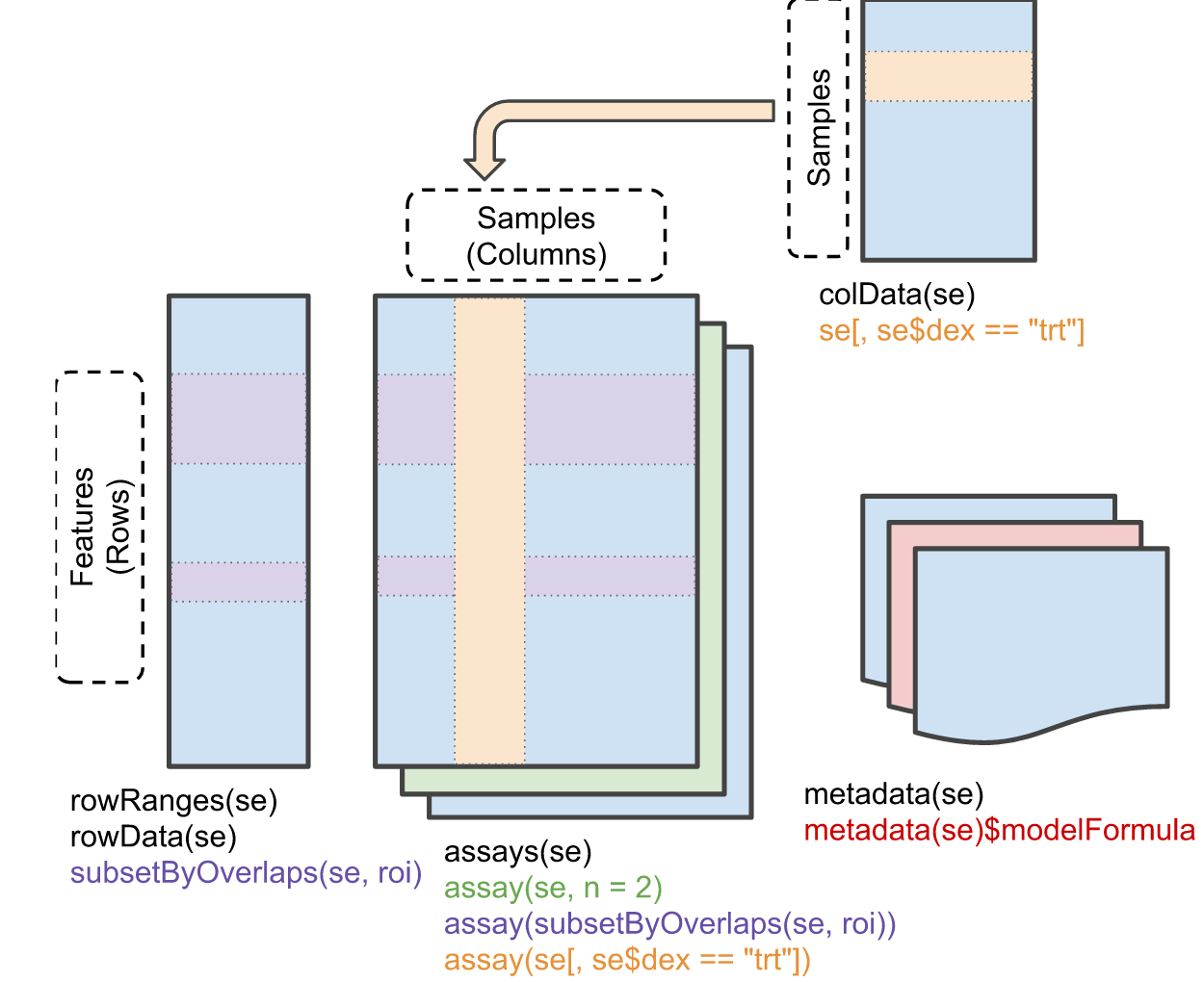
Option 3 - “The Bioconductor way”: Combine main data & metadata into one specially formatted object
Pros:
Unambiguous: keeps all data in one object with special slots that can be accessed with handy functions
Plays nice with Bioconductor tools
Efficient storage (no duplication of information like tidy way)
Cons:
Specific to Bioconductor
Not a compatible format for visualization (e.g. ggplot2)
Overall: recommended for downstream analysis (e.g. Differential Expression)
![]()
S4 is an R-specific form of Object-Oriented Programming
SummarizedExperiment: One example (there are many!) of a special object format that is designed to contain data & metadata
Comes along with handy accessor functions
Related / similar types of objects for specialized data types: RangedSummarizedExperiment, SingleCellExperiment, DGEList
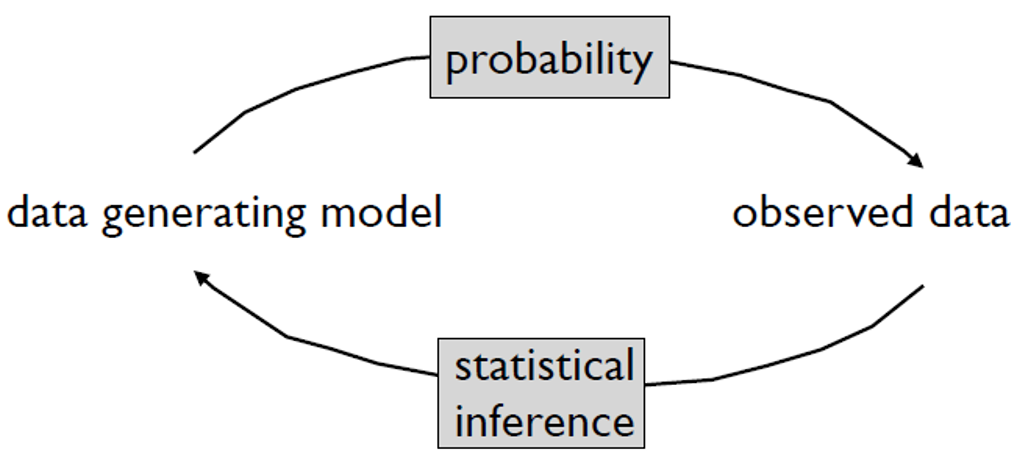
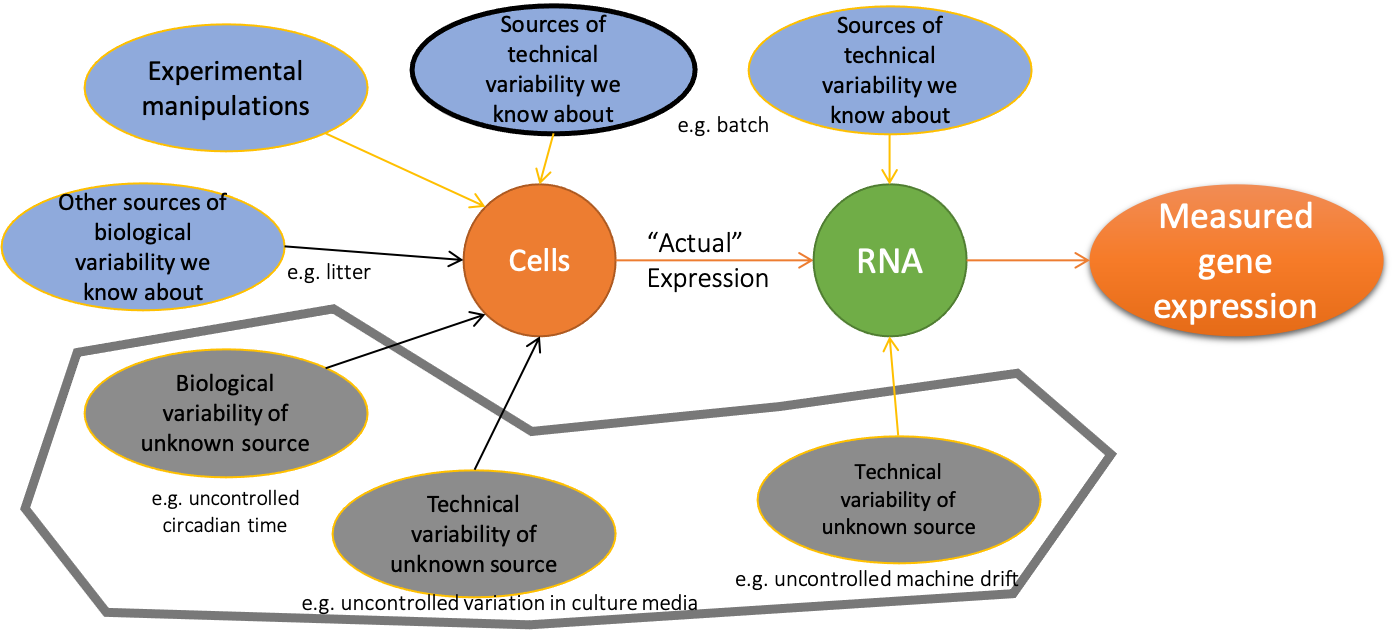
The measured expression level of gene \(g\) is the combination of many effects
Analysis goal is often to determine relative role of effects - separate signal from “noise”
If you don’t look at the data, you are likely going to miss important things
Not just at the beginning, but at every stage
That could mean making plots, or examining numerical patterns - probably both
“Sanity checks” should make up a lot of your early effort
Blindly following recipes/pipelines/vignettes/seminar code → trouble
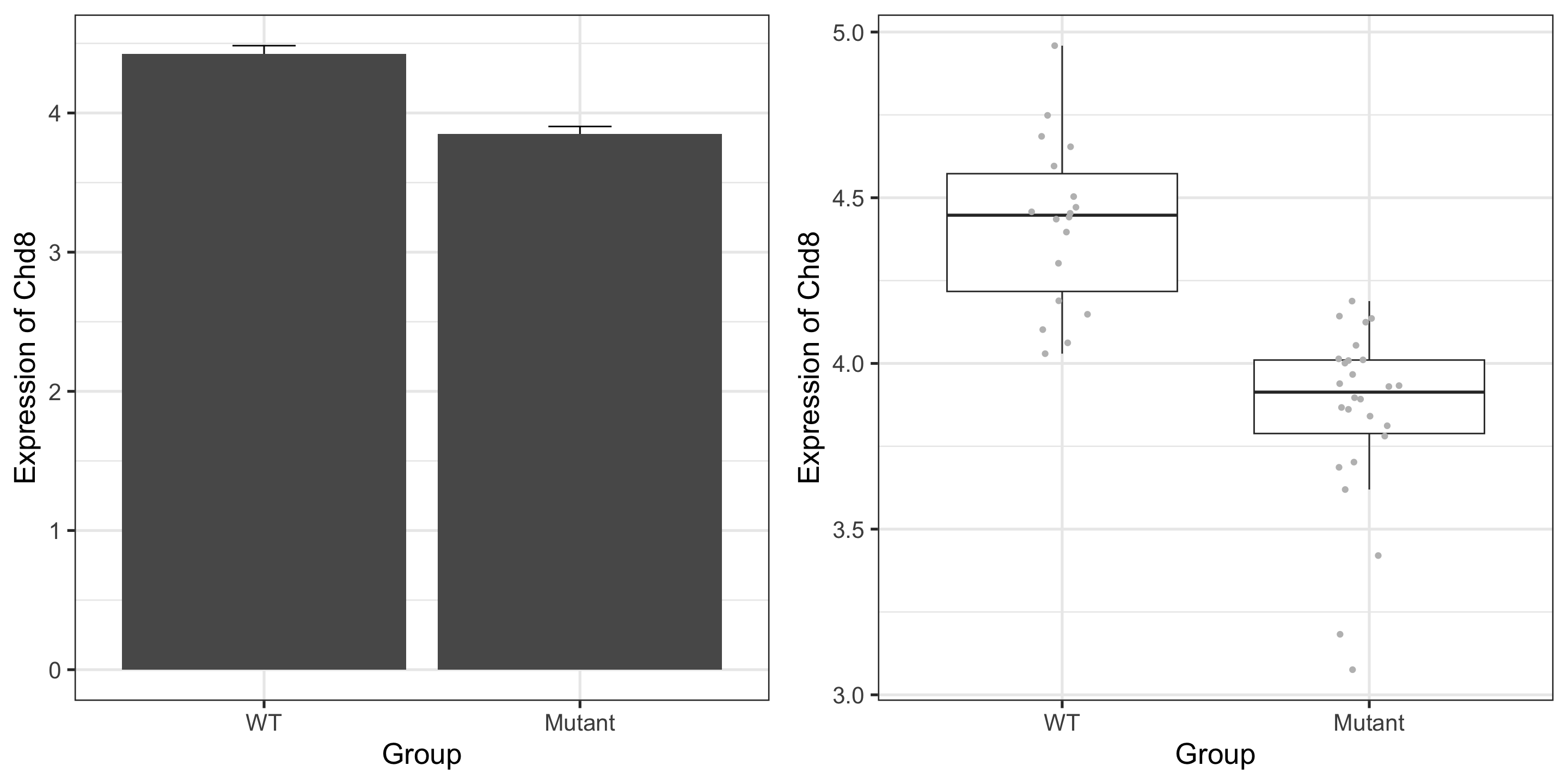
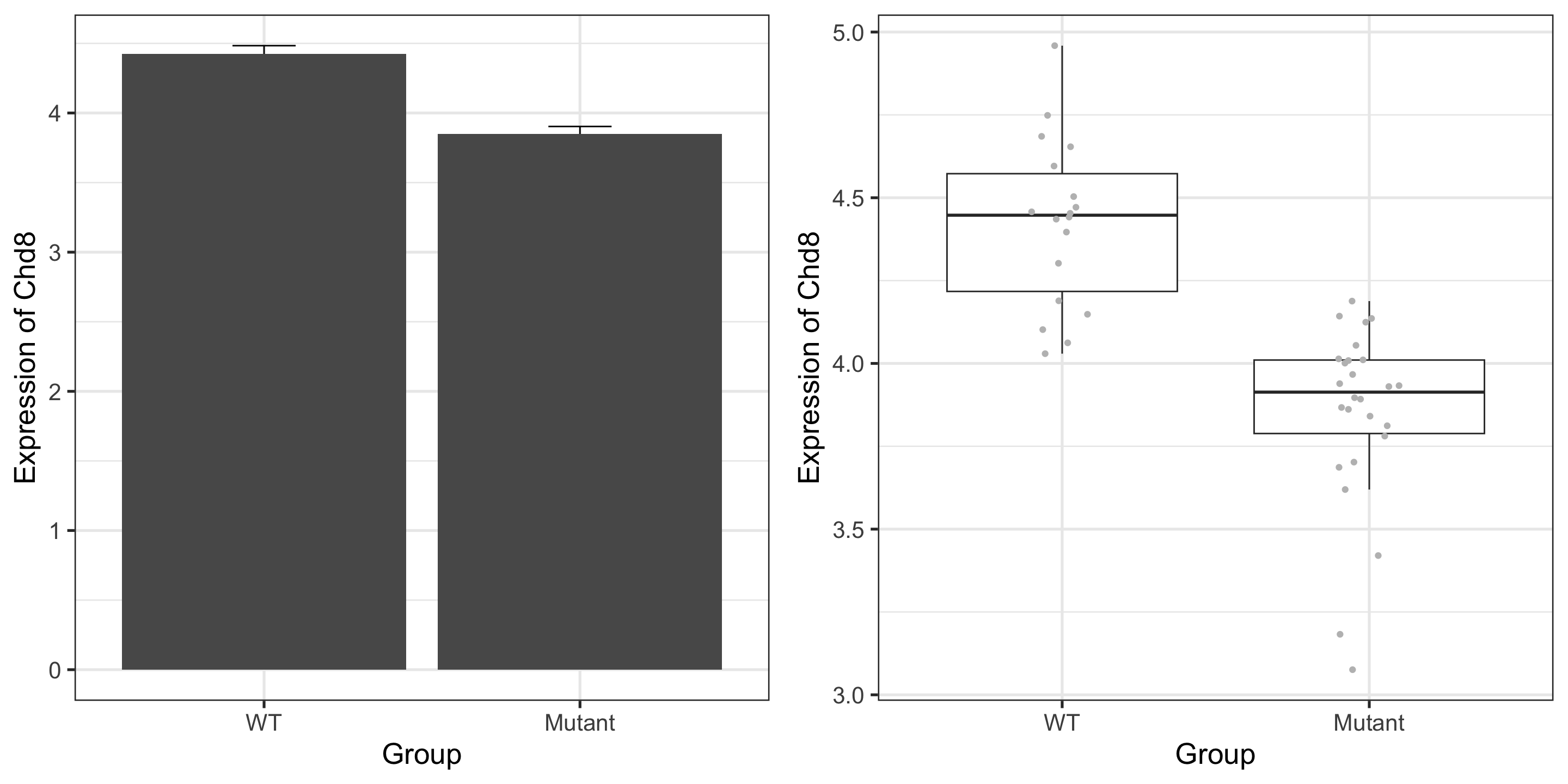
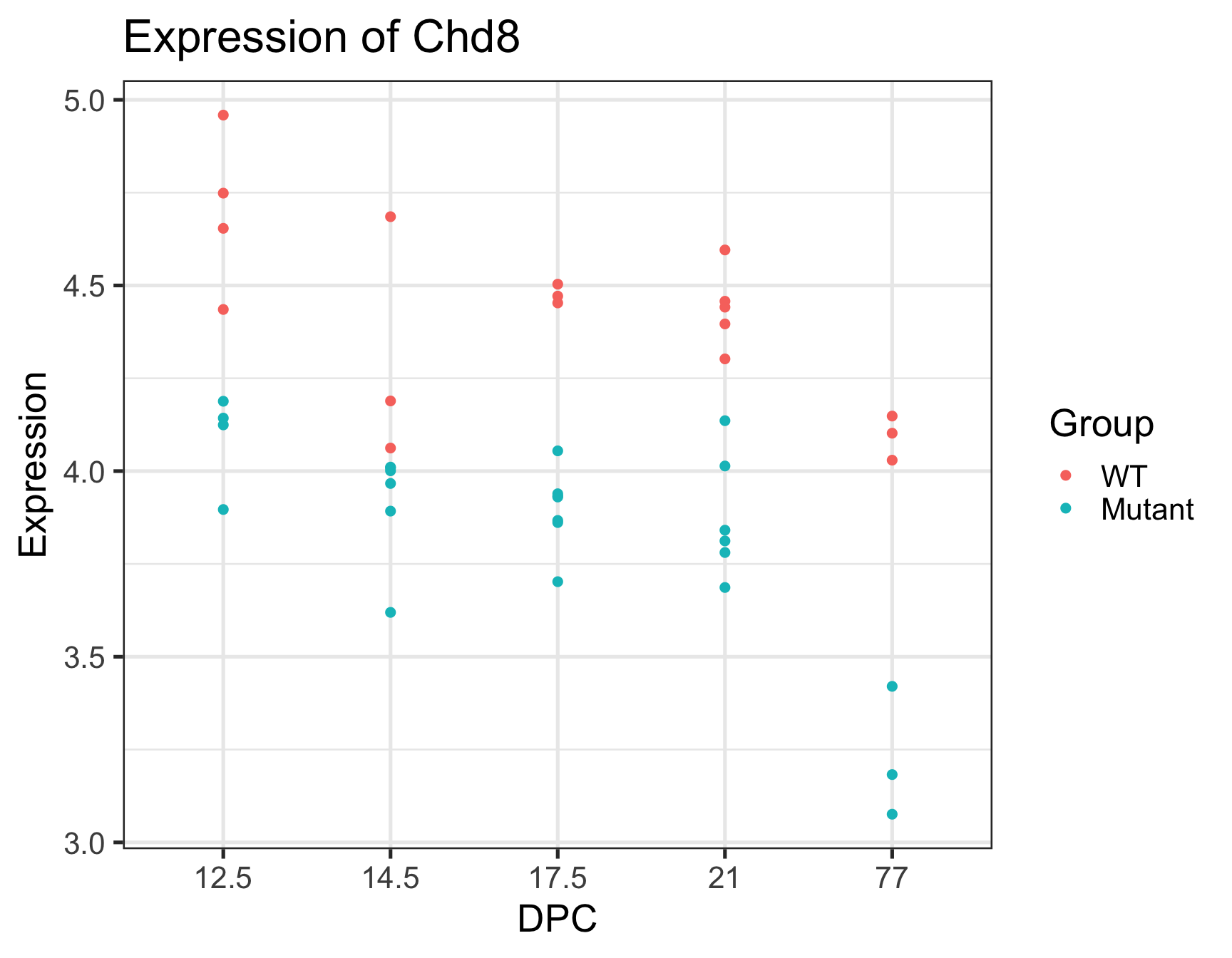
Paper reported that CHD8 went down over time, and is lower in the mutant: confirmed!
Note that we are not doing any formal “analysis” here nor trying to make this plot beautiful – keeping it very simple for our exploration
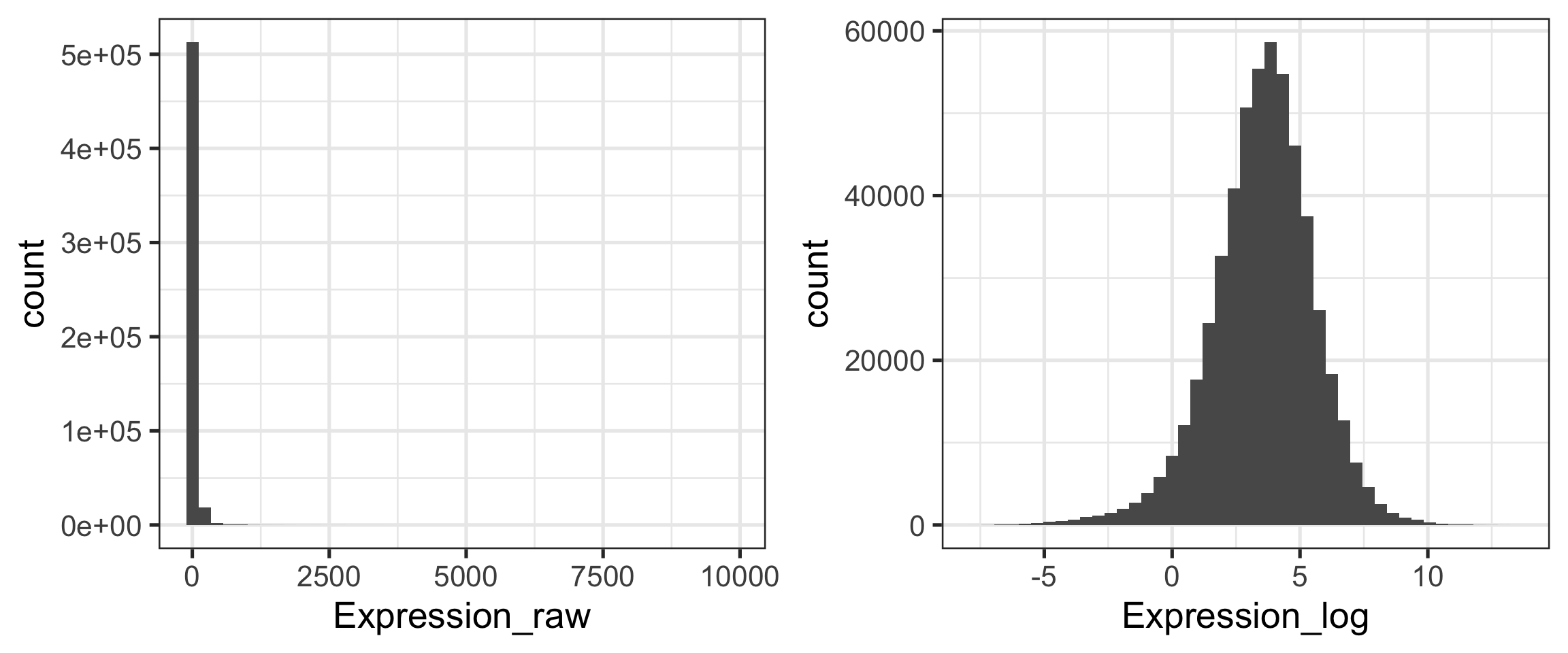
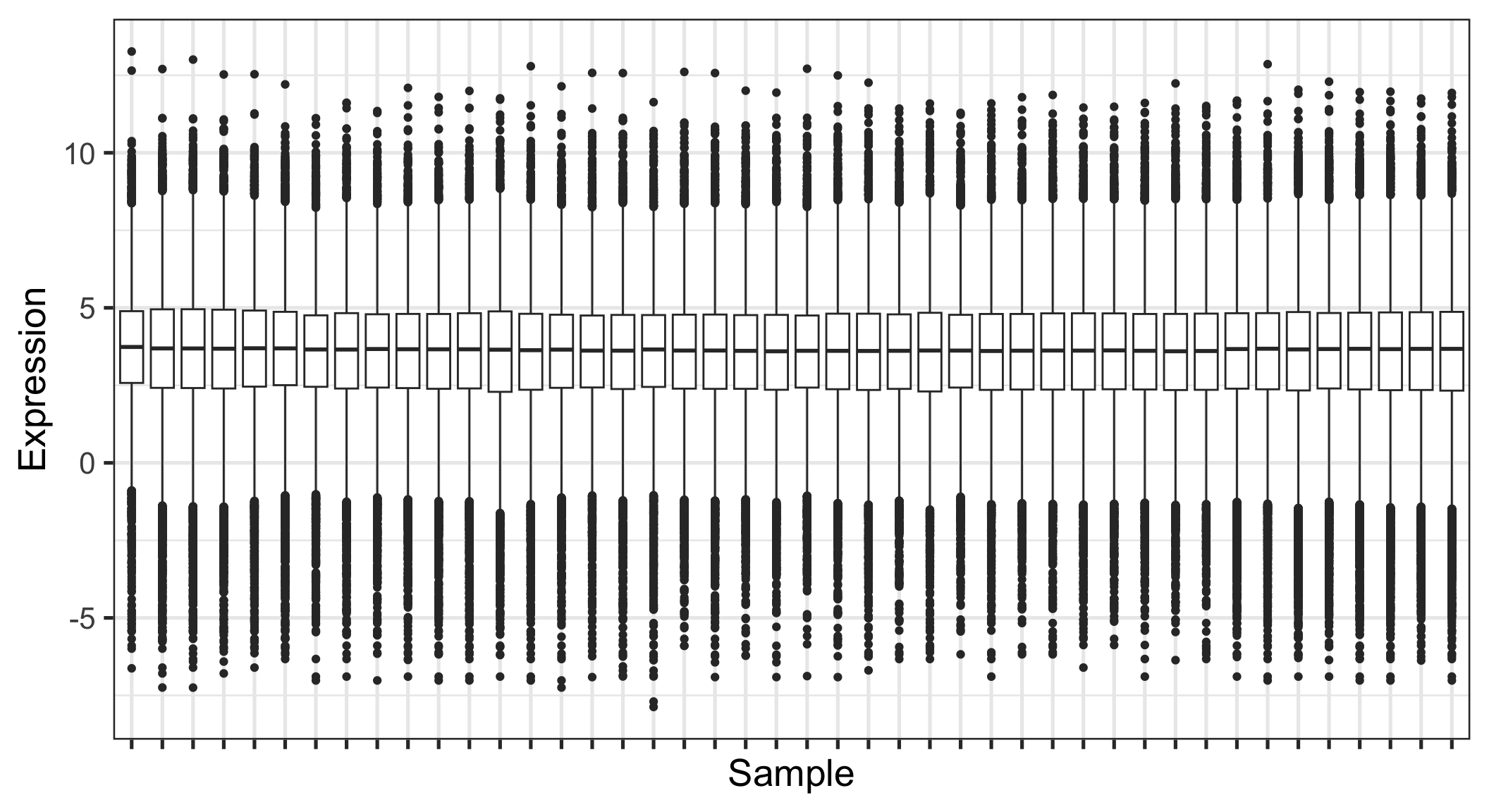
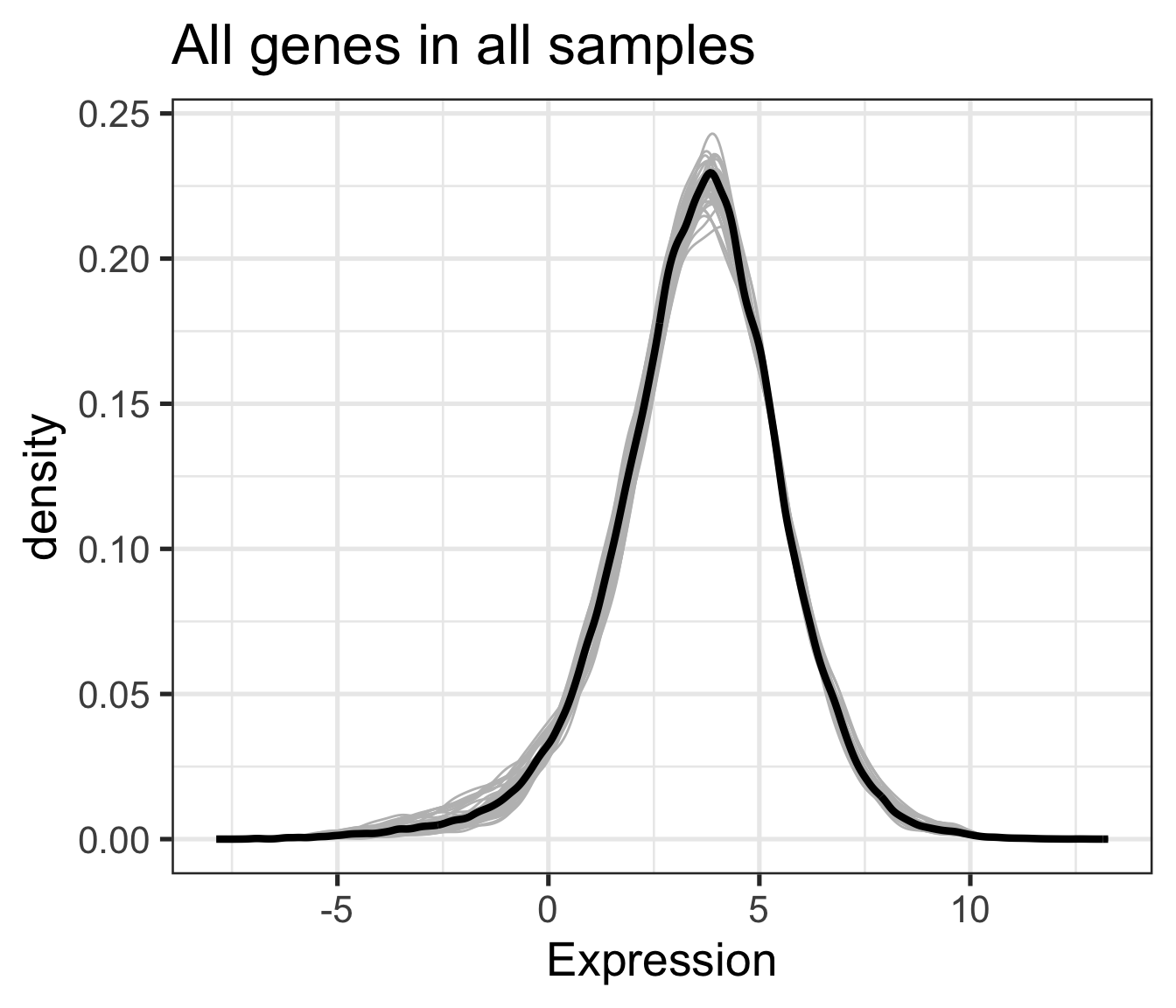
How to best summarize patterns in the data?
What is the sample size?
Is the distribution symmetrical, or skewed?
Are there any outliers?
Quick and dirty; reasonable tool to summarize large amounts of data
Not ideal if the distribution is multimodal
Don’t use box plots (alone) when you have small numbers of points; show the points!
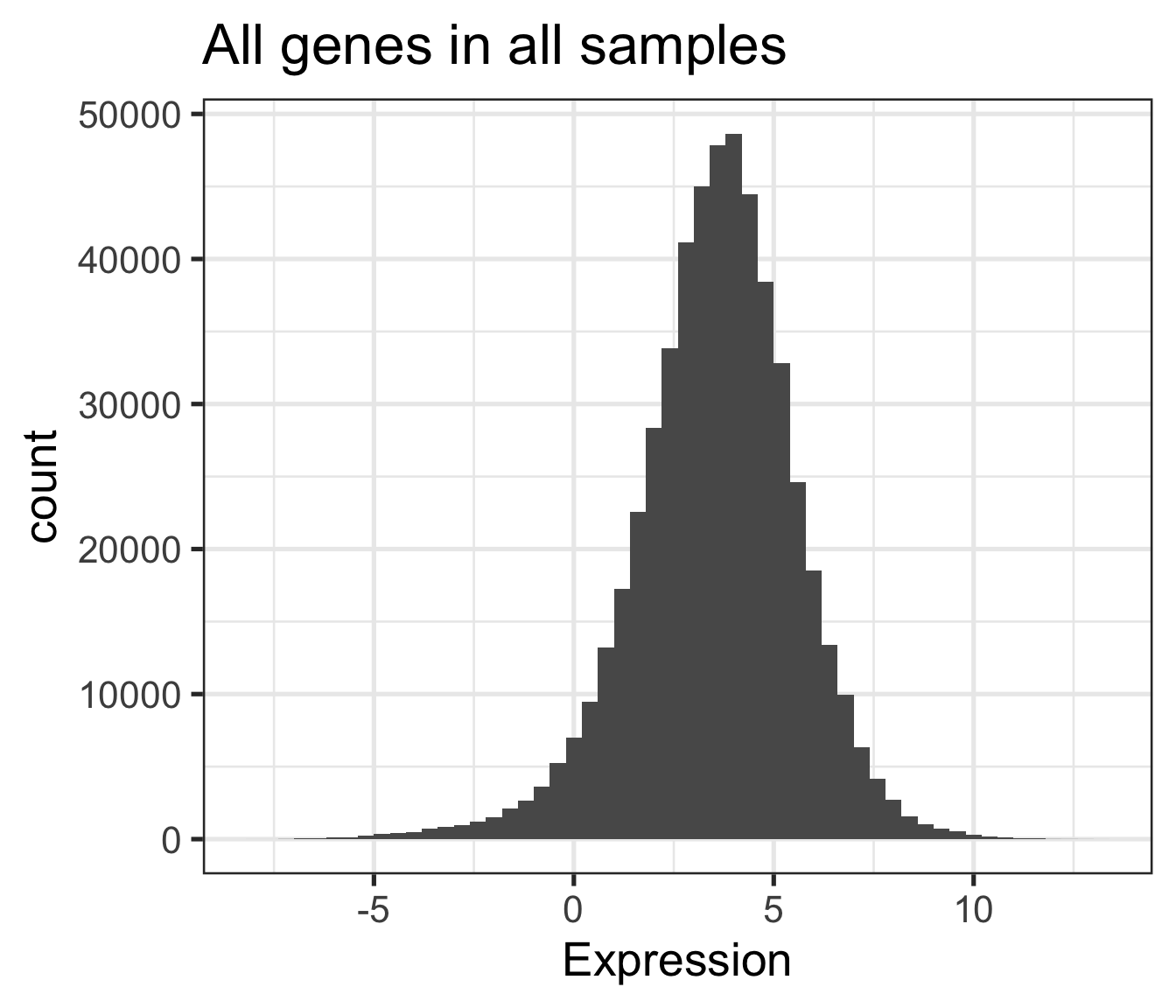
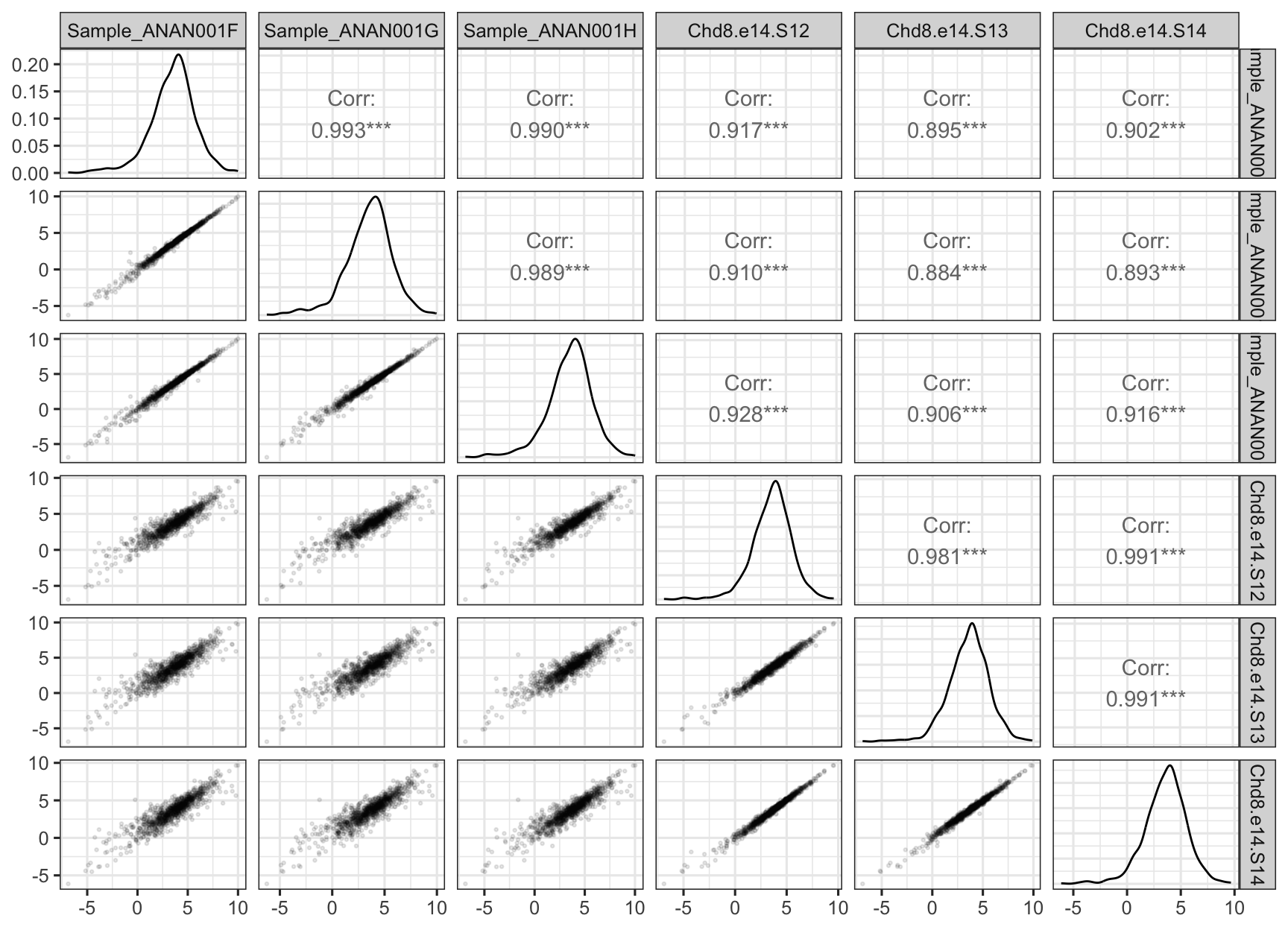
This is nice but unwieldy (or won’t work) for large data sets
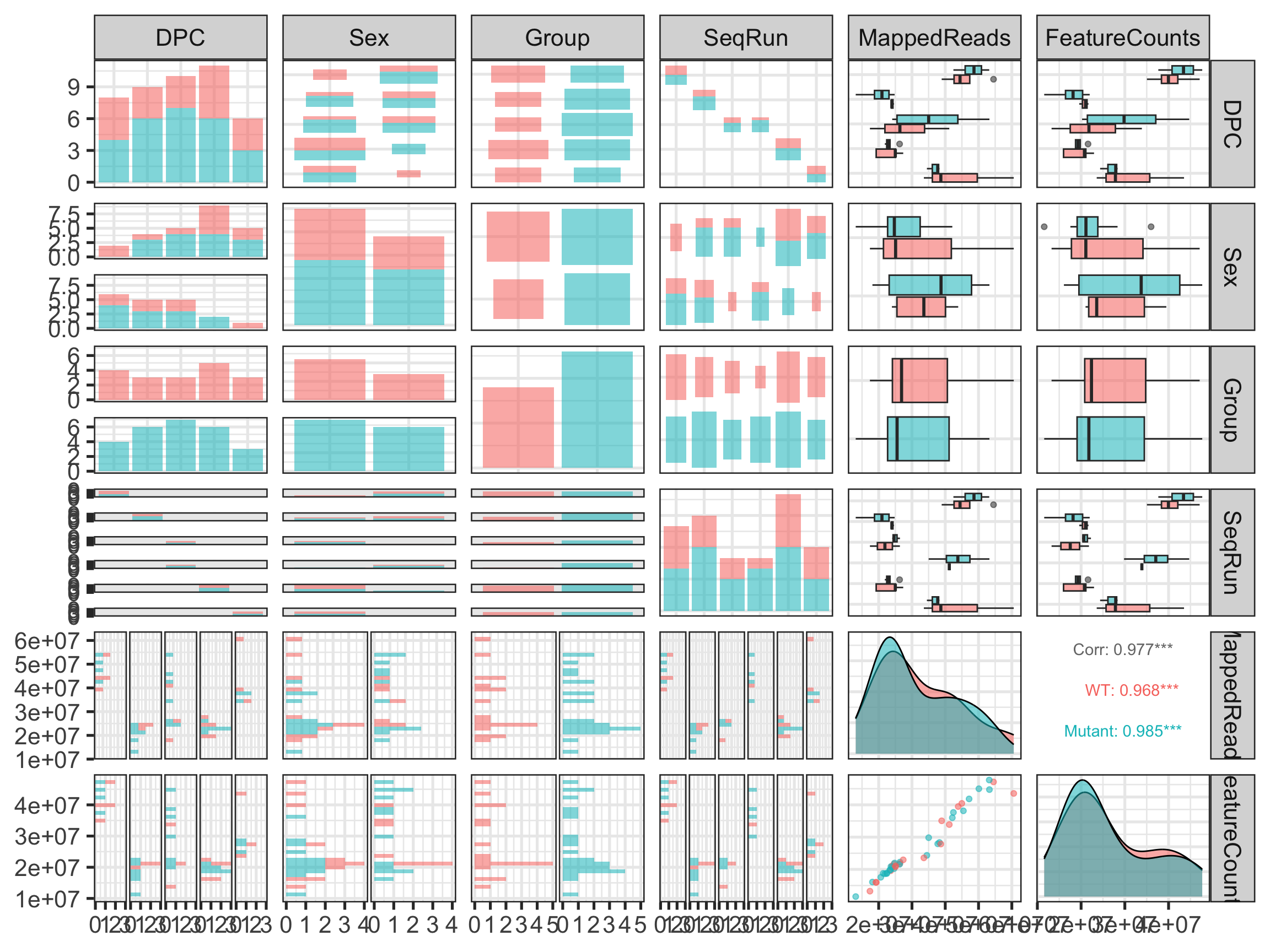
Sex is not that well balanced
There is a batch confound: The stages were run in different batches (except 17.5 was split in two)
Mapped reads varies with the batches (SeqRun)

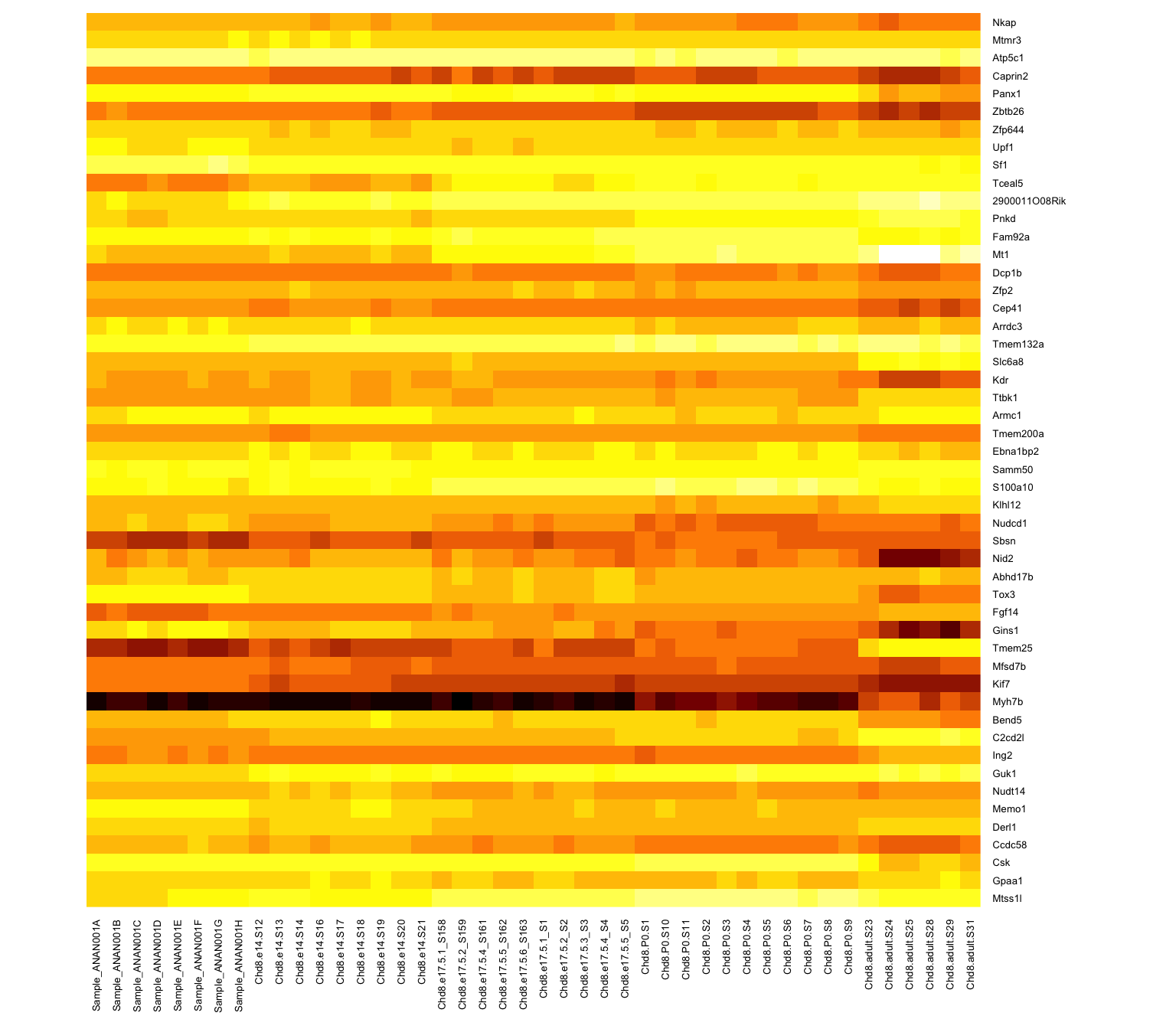
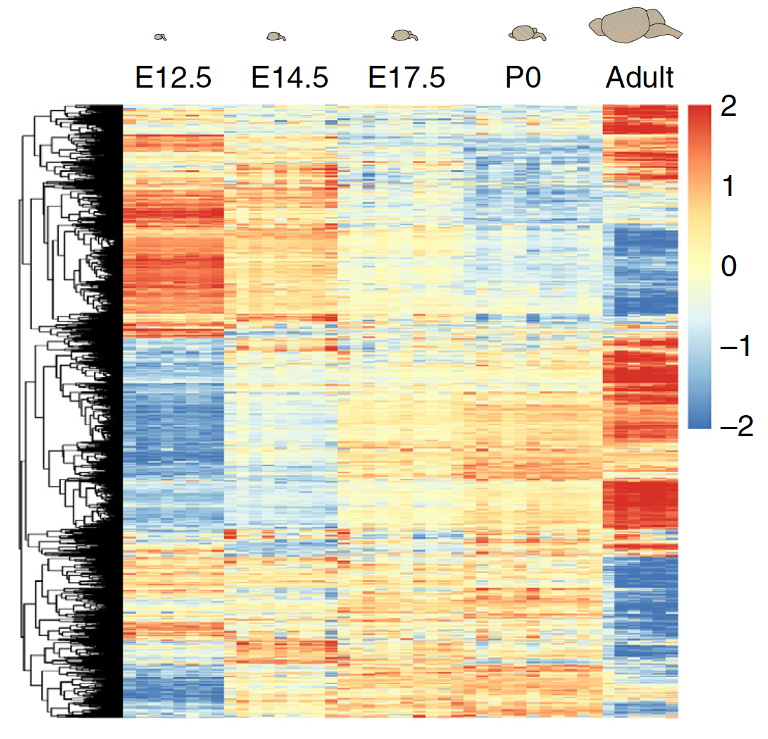
Rows are scaled to have mean 0 and variance 1 (z-scores)
Subtract the mean; divide by the standard deviation - use scale() on the data rows (some packages will do this by default)
It is now easier to compare the rows and see a bit of structure
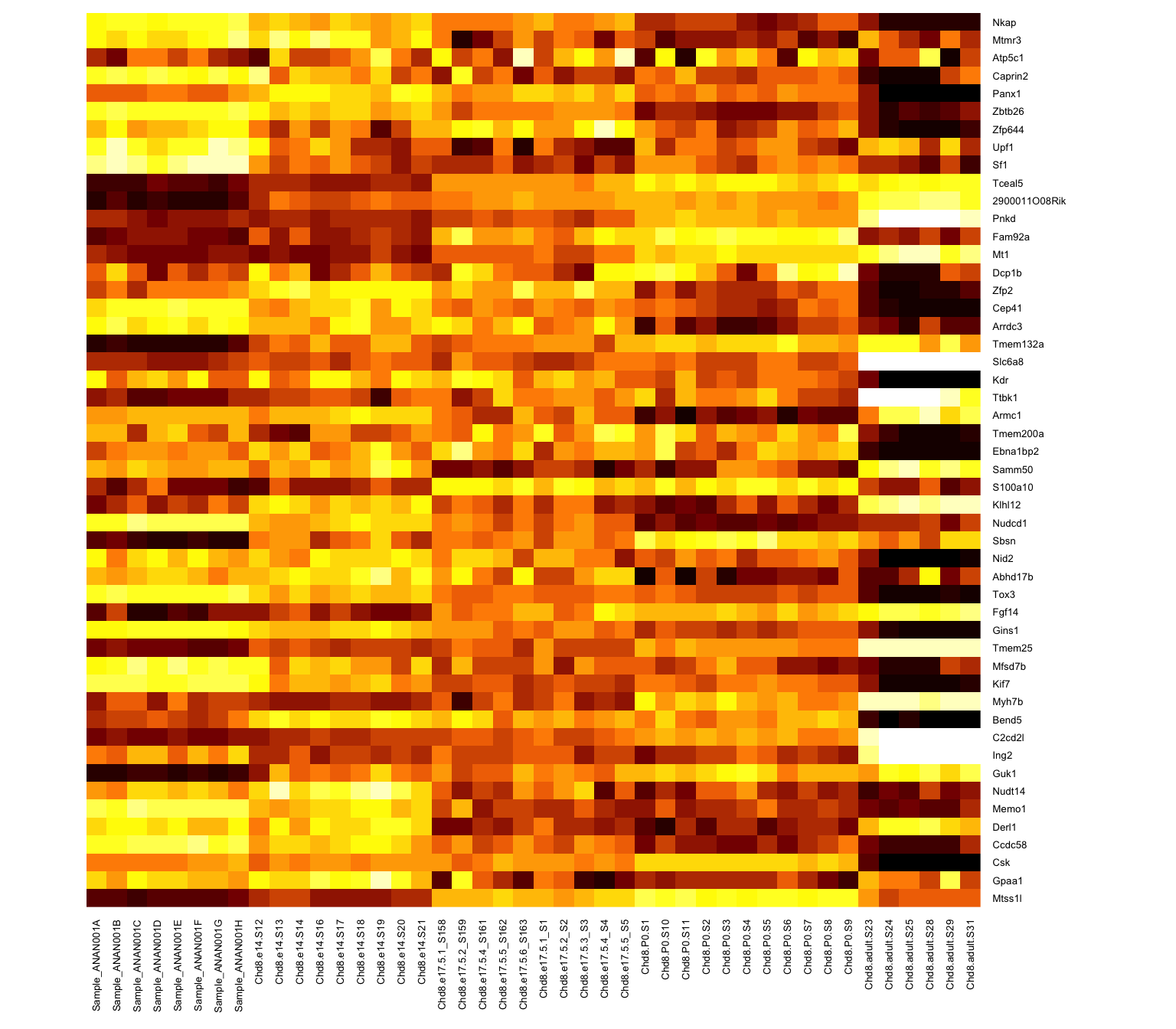
Range of values is clipped to (-2,2): aything more than two SDs from the row mean is set to 2
Limit values of 2 or 3 SDs are common
An entire data set (>10k rows)
If the cells are less than 1 pixel, everything starts to turn to mush and can even be misleading
If your heatmap has too many rows to see labels (genes), make sure it is conveying useful information (what are you trying to show?)
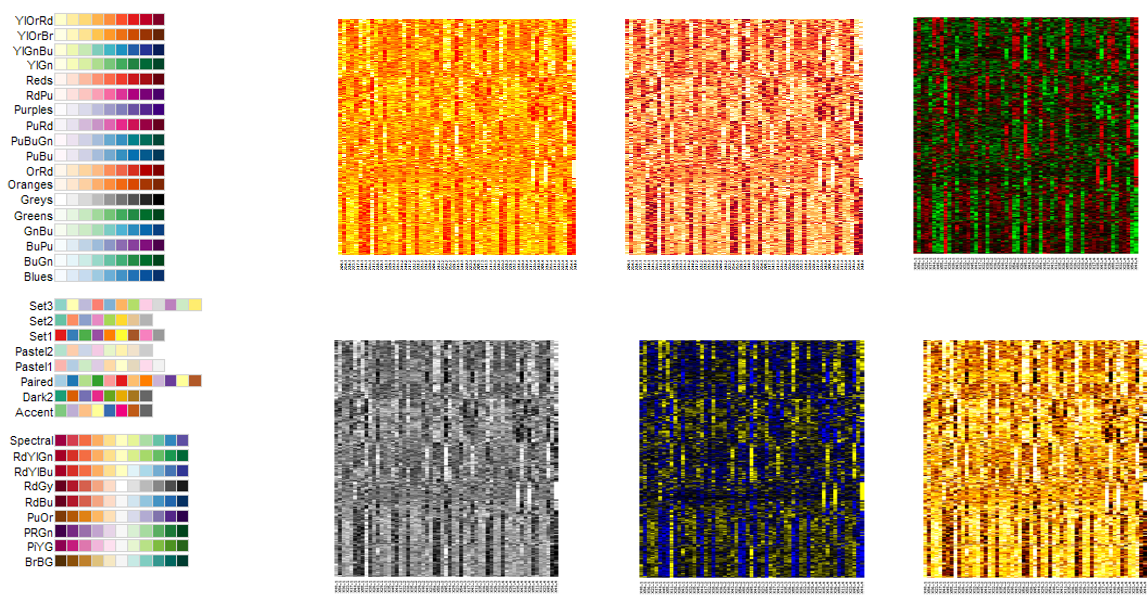
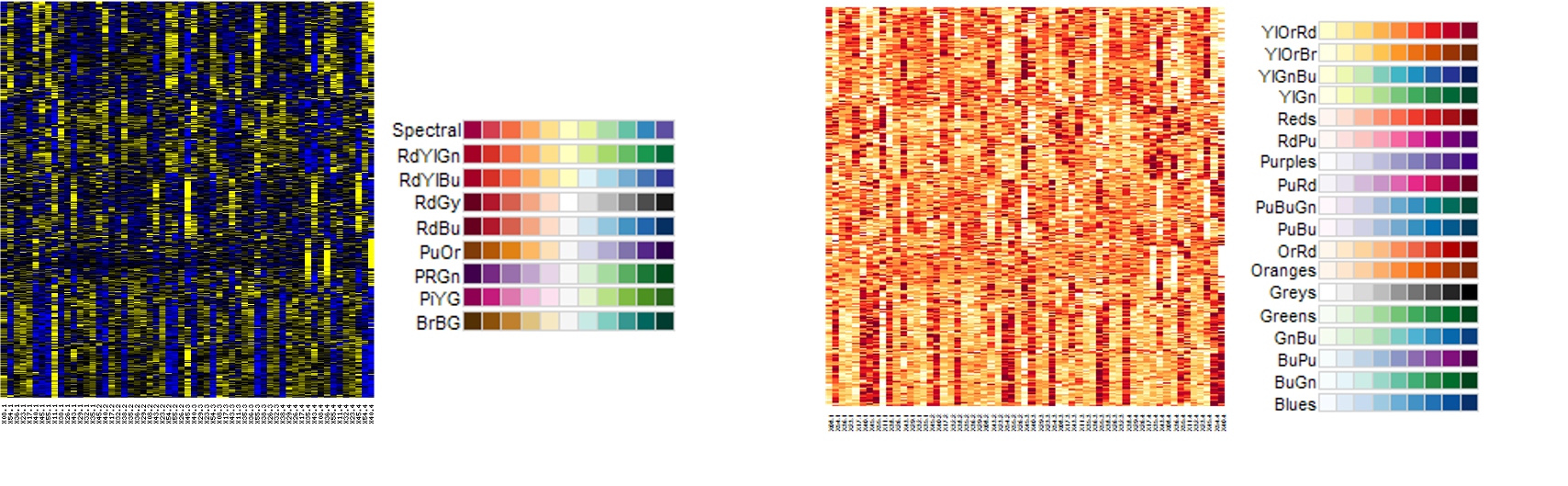
RColorBrewer: scales based on work of visualization expertsDivergent: colours pass through black or white at the midpoint (e.g. mean). Ideal if your original data are naturally “symmetric” around zero (or some other value) - Otherwise it might just be confusing
Sequential: colours go from light to dark. Darker colours mean “higher” by default in RColorBrewer. No defined midpoint.
Too many different colours to readily interpret relative ordering of values
Not recommended to use these types of scales for continuous values
Rainbows or scales with many distinct colours are better for factors / categorical variables
Pick an appropriate colour scale
Show a scale bar (so don’t use base::heatmap)
Either cluster rows / columns, or order by something meaningful (e.g. sample info)
Add annotation bars of meaningful covariates
If you have missing data points, make sure it is obvious where they are (e.g. different colour)
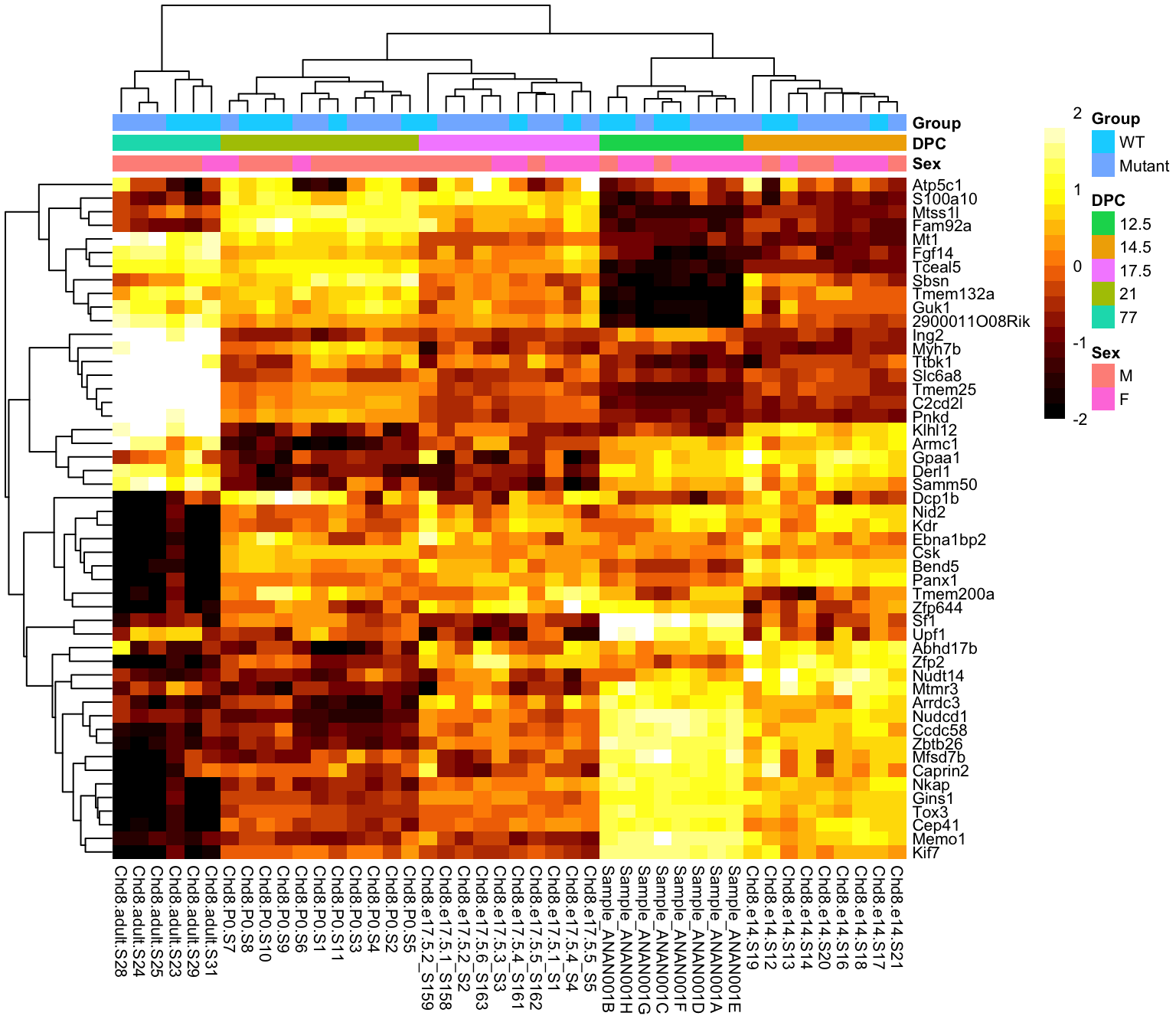
Fig 1, 10.1038/sj.ebd.6400436
Will the experiment answer the question?
Beware of (and try to control for) unwanted variation
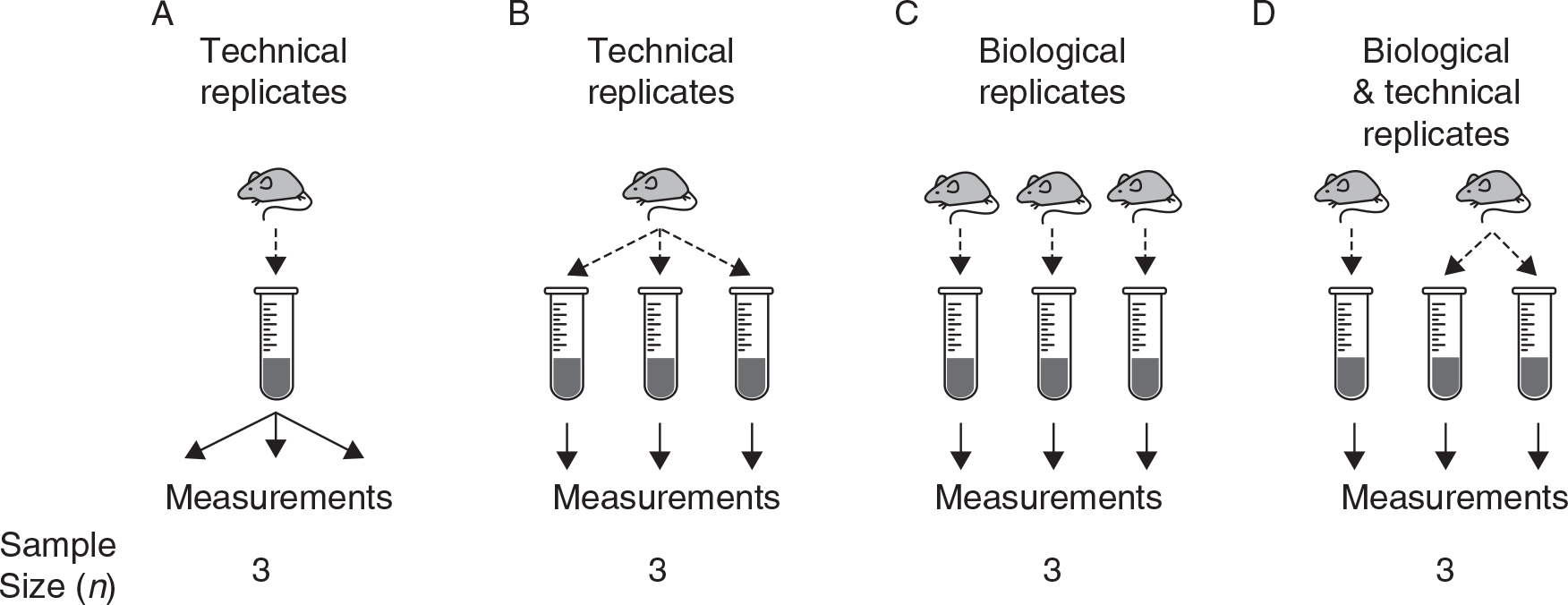
How many individuals should I study?
Biological replicates are essential and (usually) more important than technical replicates
“Batch effects are sub-groups of measurements that have qualitatively different behaviour across conditions and are unrelated to the biological or scientific variables in a study”
Definition from Leek et al. 2010 Nature Rev. Genetics 11:733
Magnitude of batch effects vary
Consider correcting for them if possible (e.g. include as covariates in model)
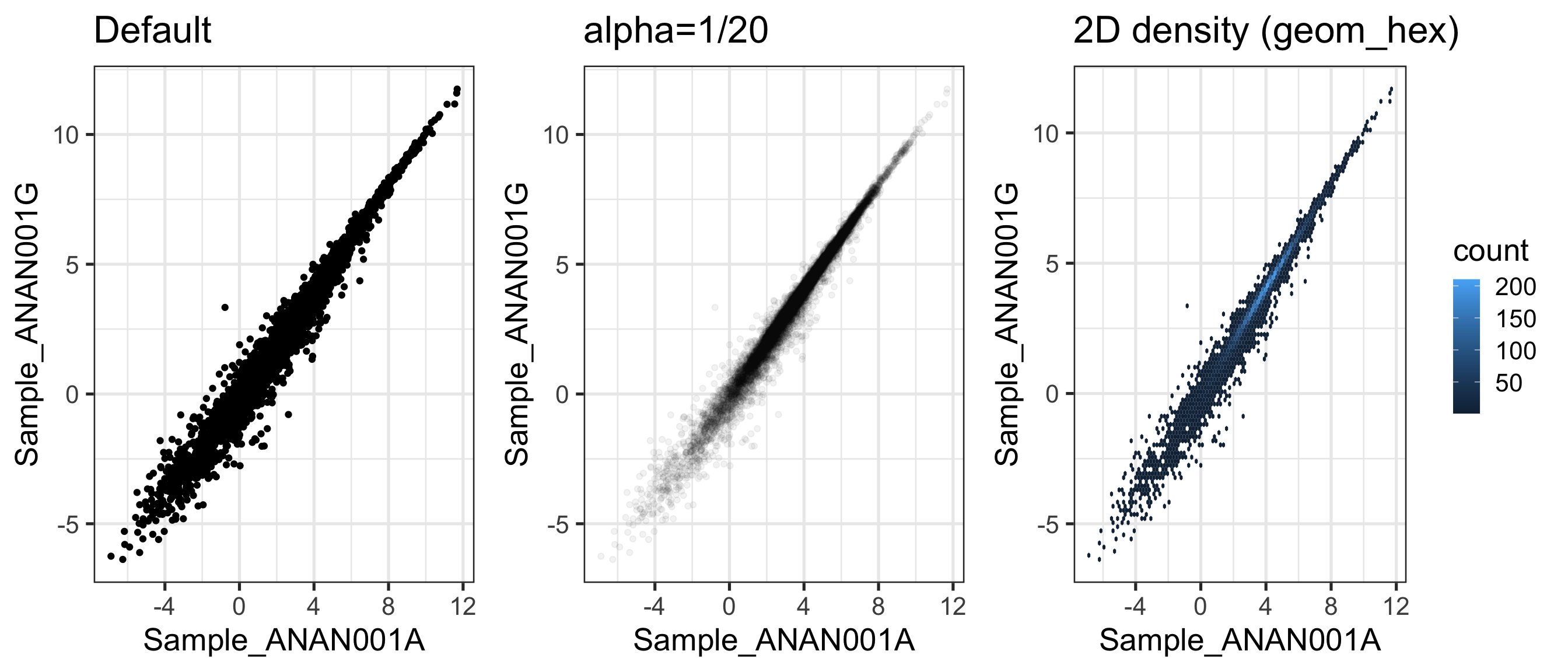
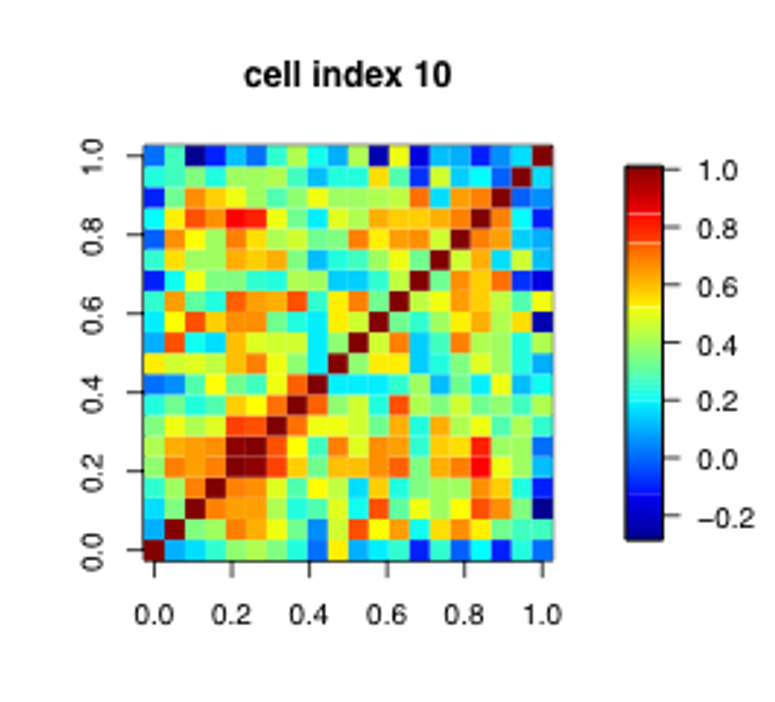
A heatmap of the sample-sample correlation matrix can help identify potential outliers
Expect correlations to be tighter within experimental groups than across groups
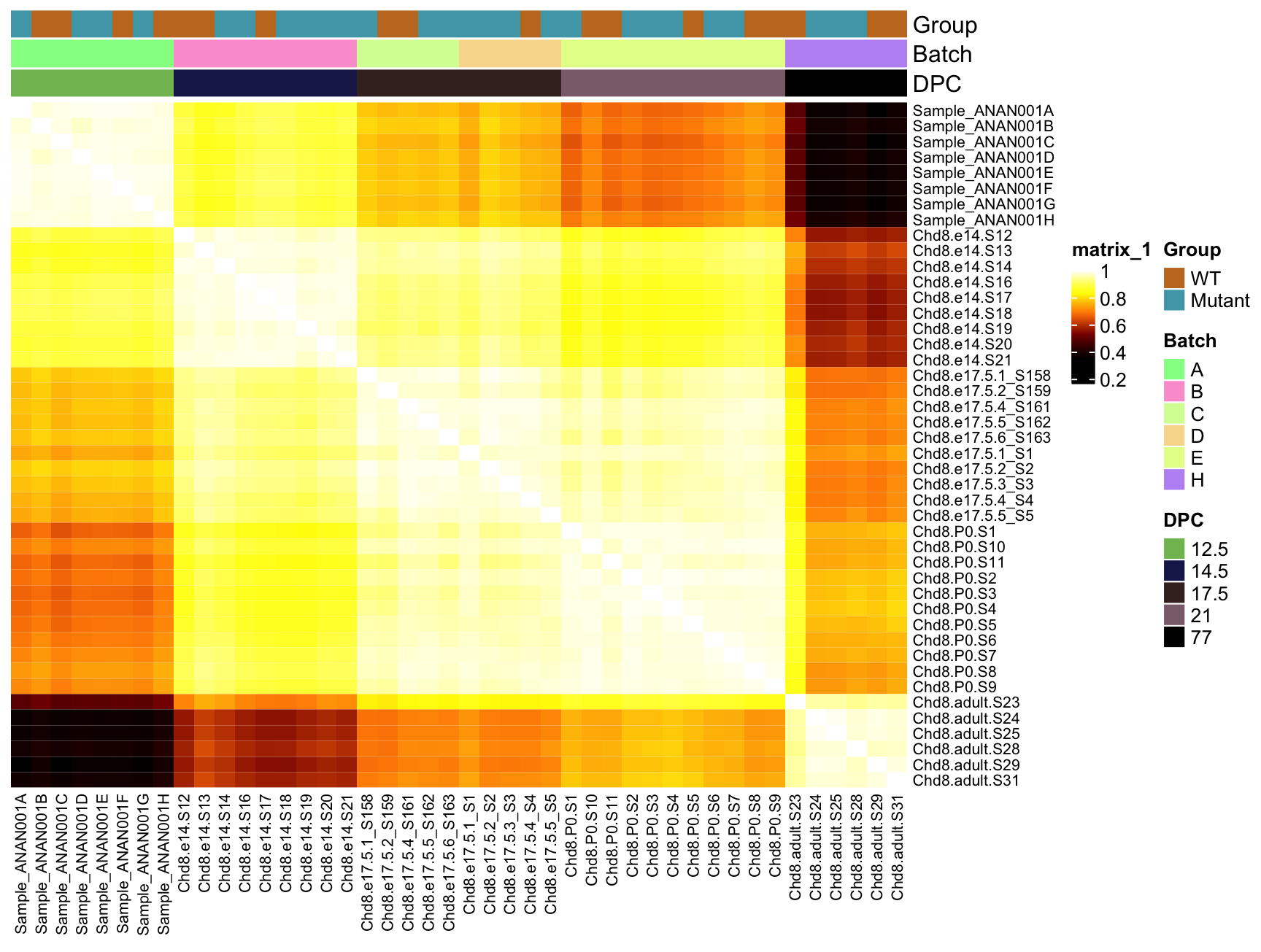
Here I mean “Removing part of the data from a sample” and doing that to all samples
In many studies, especially gene expression, it is common to remove genes that have no or very low signal (“not expressed”)
Deciding what to remove is often not straighforward, but make a principled decision and stick with it (see next slide)
Filters must be “unsupervised”
Filtering strategy should treat all samples the same
Filtering strategy should be decided up front