Recall that if \(Var(Y_i)=\sigma_Y^2\), then \(Var(\bar{Y})=\frac{\sigma_Y^2}{n_Y}\)
Assume that the random variables within each group are independent and identically distributed (iid), and that the groups are independent. More specifically, that
\(Y_1, Y_2,..., Y_{n_Y}\) are iid,
\(Z_1, Z_2,..., Z_{n_Z}\) are iid, and
\(Y, Z\) are independent.
Then, it follows that \(Var(\bar{Z}-\bar{Y})=\frac{\sigma_Z^2}{n_Z}+\frac{\sigma_Y^2}{n_Y}\)
If we also assume equal population variances: \(\sigma_Z^2=\sigma_Y^2=\sigma^2\), then \[Var(\bar{Z}-\bar{Y})=\frac{\sigma_Z^2}{n_Z}+\frac{\sigma_Y^2}{n_Y}=\sigma^2\left[\frac{1}{n_Z}+\frac{1}{n_Y}\right]\]
Reflect
Stop!
But how can we calculate population variance \(\sigma\) if it is unknown?
…using the ______________ (combined, somehow)!
# calculate sample variance for each gene and genotypetwoGenes %>%group_by(gene, genotype) %>%summarize(groupVar =var(Expression))
Unwilling to assume that F and G are normal distributions?
But you feel that nY and nZ are large enough?
Then the t-distributions above (or even a normal distribution) are decent approximations
Review
Why could we assume the sampling distribution of \(T\) is normally distributed when we have a large sample size?
Student’s t-distribution
Summary: \(T=\frac{\bar{Z}_n-\bar{Y}_n}{\sqrt{\hat{Var}(\bar{Z_n}-\bar{Y_n})}}\) is a random variable, and under certain assumptions, we can prove that \(T\) follows a t-distribution
Recall that the t-distribution has one parameter: df = degrees of freedom
Hypothesis testing: Step 1
1. Formulate your hypothesis as a statistical hypothesis
The t.test function also computes the p-value for us
In other words, assuming that H0 is true:
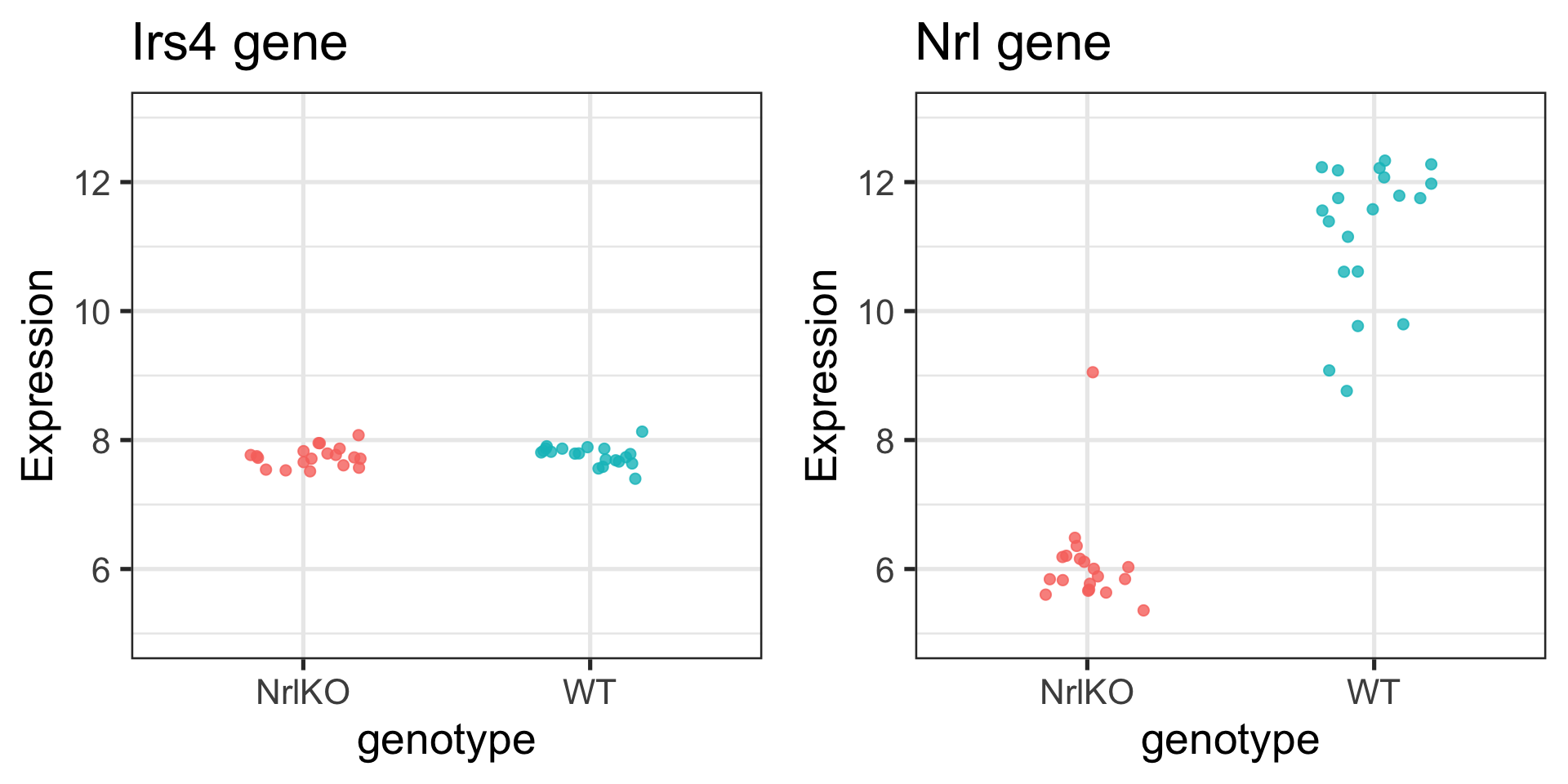
For Irs4, the probability of seeing a test statistic as extreme as that observed \((t = -0.53)\) is pretty high \((p = 0.6)\).
But for Nrl, the probability of seeing a test statistic as extreme as that observed \((t = -16.8)\) is extremely low \((p=6.76 \times 10^{-19})\)
Hypothesis Testing: Step 4
4. Make a decision about significance of results
The decision should be based on a pre-specified significance level (\(\alpha\))
\(\alpha\) is often set at 0.05. However, this value is arbitrary and may depend on the study.
Irs4
Using \(\alpha=0.05\), since the p-value for the Irs4 test is greater than 0.05, we conclude that there is not enough evidence in the data to claim that Irs4 has differential expression in WT compared to NrlKO models.
We do not reject H0!
Nrl
Using \(\alpha=0.05\), since the p-value for the Nrl test is much less than 0.05, we conclude that there is significant evidence in the data to claim that Nrl has differential expression in WT compared to NrlKO models.
We reject H0!
t.test function in R
Assuming equal variances
twoGenes %>%filter(gene =="Nrl") %>%t.test(Expression ~ genotype, var.equal=TRUE, data = .)
Two Sample t-test
data: Expression by genotype
t = -16.798, df = 37, p-value < 2.2e-16
alternative hypothesis: true difference in means between group NrlKO and group WT is not equal to 0
95 percent confidence interval:
-5.776672 -4.533071
sample estimates:
mean in group NrlKO mean in group WT
6.089579 11.244451
Not assuming equal variances
twoGenes %>%filter(gene =="Nrl") %>%t.test(Expression ~ genotype, var.equal=FALSE, data = .)
Welch Two Sample t-test
data: Expression by genotype
t = -16.951, df = 34.01, p-value < 2.2e-16
alternative hypothesis: true difference in means between group NrlKO and group WT is not equal to 0
95 percent confidence interval:
-5.772864 -4.536879
sample estimates:
mean in group NrlKO mean in group WT
6.089579 11.244451
Tip
Check out ?t.test for more options, including how to specify one-sided tests
Interpreting p-values
Which of the following are true? (select all that apply)
If the effect size is very small, but the sample size is large enough, it is possible to have a statistically significant p-value
A study may show a relatively large magnitude of association (effect size), but a statistically insignificant p-value if the sample size is small
A very small p-value indicates there is a very small chance the finding is a false positive
Common p-value pitfalls
Caution
Valid inference using p-values depends on accurate assumptions about null sampling distribution
Caution
A p-value is NOT:
The probability that the null hypothesis is true
The probability that the finding is a “fluke”
A measure of the size or importance of observed effects
Preview: “Genome-wide” testing of differential expression
In genomics, we often perform thousands of statistical tests (e.g., a t-test per gene)
The distribution of p-values across all tests provides good diagnostics/insights
Is it mostly uniform (flat)? If not, is the departure from uniform expected based on biological knowledge?
We will revisit these topics in greater detail in later lectures
Different kinds of t-tests:
One sample ortwo samples
One-sided ortwo sided
Paired orunpaired
Equal variance or unequal variance
Types of Errors in Hypothesis Testing
\[ \alpha = P(\text{Type I Error}), \text{ } \beta = P(\text{Type II Error}), \text{ Power} = 1- \beta\]
H0: “Innocent until proven guilty”
The default state is \(H_0 \rightarrow\) we only reject if we have enough evidence
If \(H_0\): Innocent and \(H_A\): Guilty, then
Type I Error \((\alpha)\): Wrongfully convict innocent (False Positive)
Type II Error \((\beta)\): Fail to convict criminal (False Negative)
Willing to assume that F and G are normal distributions?
Unwilling to assume that F and G are normal distributions?
But you feel that nY and nZ are large enough?
Then the t-distributions above (or even a normal distribution) are decent approximations
Stop!
What if we aren’t comfortable assuming the underlying data generating process is normal AND we aren’t sure our sample is large enough to invoke the CLT?
What are alternatives to the t-test?
First, one could use the t test statistic but use a permutation approach to compute its p-value; we’ll revisit this topic later
Non-parametric tests are an alternative:
Wilcoxon rank sum test (Mann Whitney) uses ranks to test differences in population means
Kolmogorov-Smirnov test uses the empirical CDF to test differences in population cumulative distributions
Wilcoxon rank sum test
Rank all data, ignoring the grouping variable
Test statistic = sum of the ranks for one group (optionally, subtract the minimum possible which is \(\frac{n_Y(n_Y+1)}{2}\))
Alternative but equivalent formulation based on the number of \(y_i, z_i\) pairs for which \(y_i \geq z_i\)
The null distribution of such statistics can be worked out or approximated