[1] 735471Gene set analysis
February 27, 2024
Biological mechanisms / processes are modular
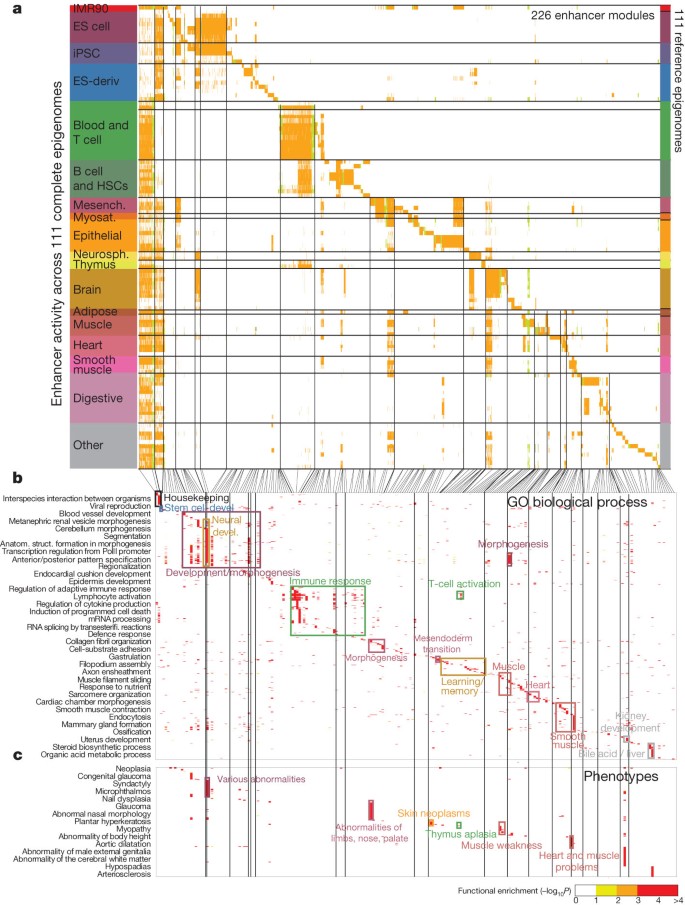
Roadmap epigenomics, Nature 2015
Epigenomic (enhancer element) modules across 111 human tissues
↓
Gene Ontology information confirms cell-type or tissue-specific biological pathways
Clusters of cells correspond to ‘cell types’
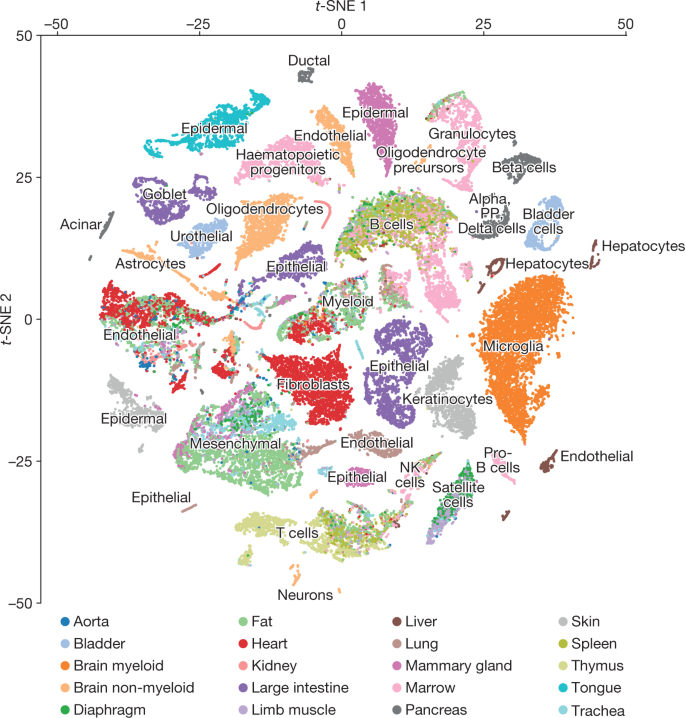
Tabula Muris, Nature 2018: “Comparison of cell-type determination as performed by unbiased whole-transcriptome comparison versus manual annotation of clusters by organ-specific experts.”

Enrichment analysis to confirm previous knowledge

Where does the ‘dictionary’ come from?
Gene Ontology (GO) is a controlled vocabulary for function
Began ~25 years ago; over 45K terms today
Mostly species-agnostic
Three aspects:
- Biological Process
- Cellular Component
- Molecular Function
![]()

GO structure
- Structure is an acyclic graph, not a tree: terms commonly have multiple parents
- Annotations in the database don’t separately list out all the parents
- Includes a mix of ‘directly annotated’ and many ‘inferred’ terms
- Several relationship types


Pathways: Another grouping of genes

Example: Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathways database
Species-specific directed networks of genes and their relationships, curated from the literature
Pathways are organized into functional categories (not nearly as complex as GO)
What have we learned from our differential expression analysis?

blue: some biological pathway of interest
Let’s drop the edges in biological networks

Prior Knowledge of biological pathways define a set of genes (blue)
Does this pathway overlap with our DEG list?

Suppose we discovered the 8 DEG outlined (pink)
Is our DEG list enriched for genes in this pathway?

Suppose we found \(q\) genes are overlapping with this pathway (green)
Gene Set Analysis testing over-representation of DEGs

What are the relevant numbers?
\(N\): # genes in this universe
\(m\): # genes in this set
\(n\): # genes not in this gene set
\(k\): # DEGs in our analysis
\(q\): # DEGs (of \(k\)) overlapping with the set of \(m\) genes
Is this overlap of \(q=6\) of \(k=8\) genes significant?
In this example:
\(N = 24\) total genes
\(m = 9\) genes in this set
\(n = N - m = 15\) not in this set
\(k = 8\) DEGs
\(q=6\) out of \(k=8\) overlap
Is this overlap of \(q=6\) of \(k=8\) genes significant?
Questions:
Is it meaningful enough to report?
Is it surprising enough that we recapitulated 6/9 (~67 %) genes in the set?
Is it surprising enough that we 6/8 (~75 %) of our DEG are in the set?
What is the null distribution?
What is the generative/simulation scheme?
How do we find \(q\) out of \(k\) DEGs overlapping with a gene set of \(m\) genes?
Under the null of hypergeometric distribution
Randomly sample \(k\) DEGs out of \(N\) genes
Of these \(k\) genes, \(q\) overlap with a gene set consisting of \(m\) genes
The rest \(k-q\) genes overlap with genes outside of the gene set \(N-m\)
How to generalize?
Probability distribution to describe this process
Hypergeometric distribution
A discrete probability distribution that describes the probability of \(q\) successes (random draws for which the object drawn has a specified feature) in \(k\) draws, without replacement, from a finite population of size \(N\) that contains exactly \(m\) objects with that feature.

image source: en.wikipedia.org/wiki/Hypergeometric_distribution
Hypergeometric distribution
Probability mass function
\[ \begin{eqnarray*} P(x=q|N,m,k) &=& \sum_{\begin{array}{l} {\color{blue} \textsf{# ways to select }q\textsf{ out of }m } \\ {\color{teal} \textsf{# ways to select }(k-q)\textsf{ out of }N-m } \end{array}} {\color{magenta} \begin{array}{l} \textsf{the probability} \\ \textsf{of choosing a set size }k \\ \textsf{out of total }N \end{array}} \\ &=& { \underbrace{\color{blue} {m \choose q}}_{ \begin{array}{l} {\color{blue}\textsf{# ways to choose }} \\ {\color{blue}q\, \textsf{ overlap out of }m} \end{array} } } { \times \underbrace{\color{teal} {N-m \choose k - q}}_{ \begin{array}{l} {\color{teal}\textsf{# ways to choose }} \\ {\color{teal}(k-q) \textsf{ out of }N-m} \end{array} } } { \times {\color{magenta} {N \choose k}^{-1} } } \end{eqnarray*} \]

How significant is \(q\) overlap in our discovery?
\(m=9\), \(n=15\), & \(k=8\):

If \(q=3\):

If \(q=6\):

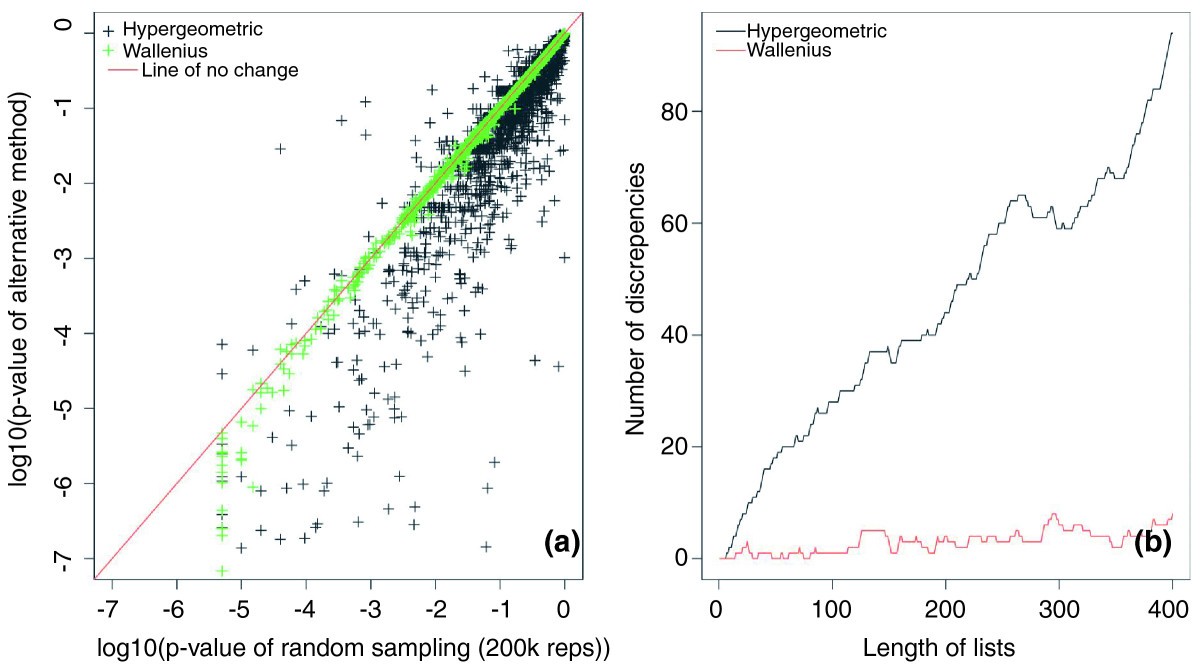
Not all gene sets are the same (gene length bias)
So far, we assumed all genes are independent and equally likely to be DE under the null

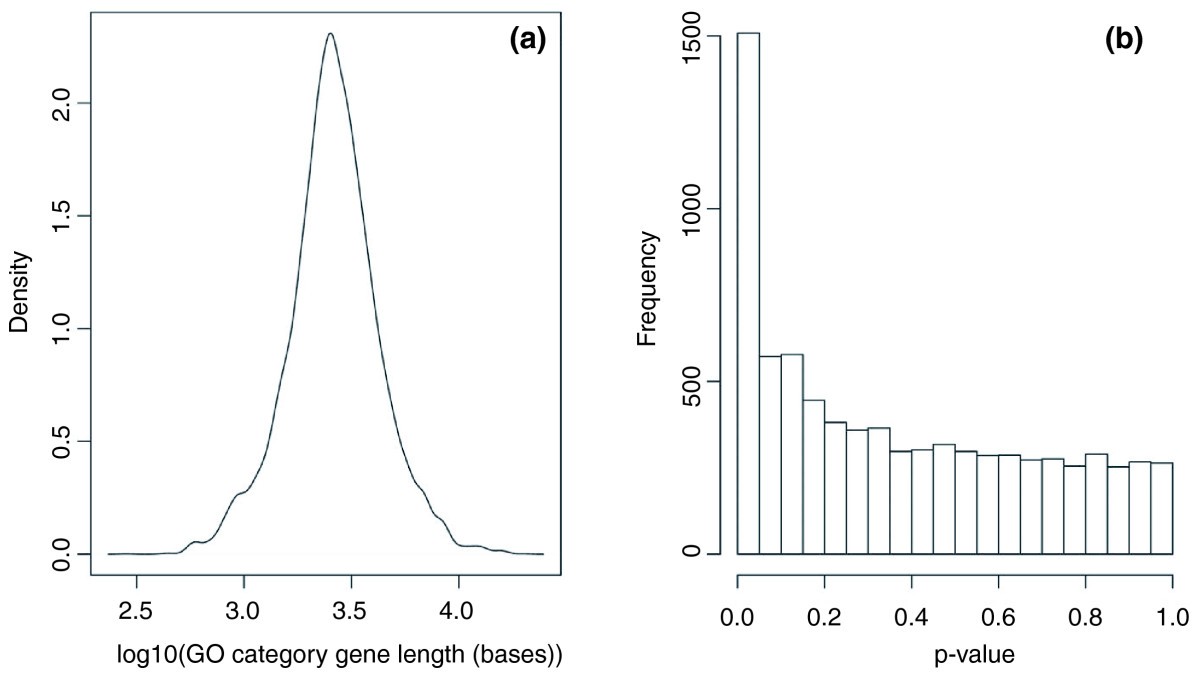
Distribution of average gene lengths in GO categories
Wilcoxon p-values of median length of genes in 7,873 GO categories compared to overall
Longer genes tend to be more differentially expressed
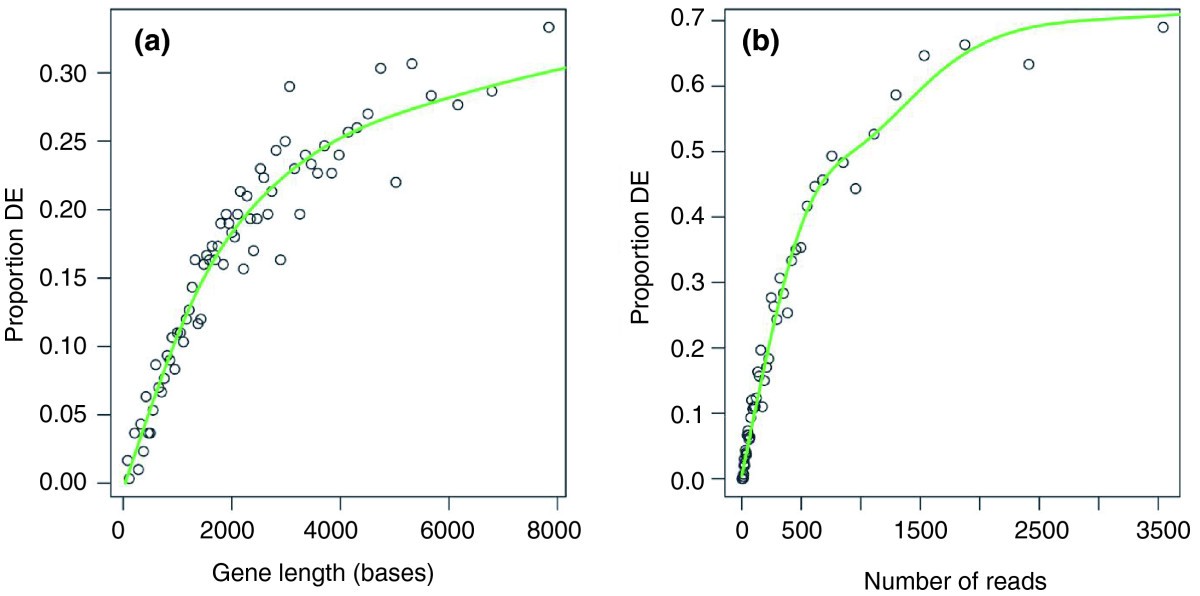
GOseq null distribution conditions on gene lengths
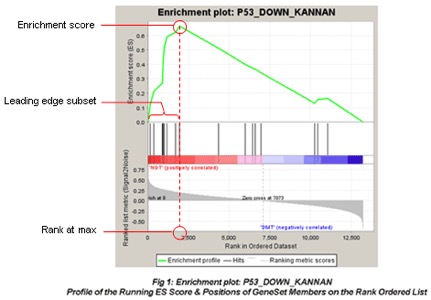
Rank-based Gene Set Enrichment Analysis (GSEA)

Rank-based GSEA (Subramanian et al. 2005)
A collection of gene-sets: \(\mathcal{C}_{1}, \ldots, \mathcal{C}_{K}\)
A vector of gene-level scores (\(G\) genes): \(z_{1}, \ldots, z_{G}\)
Each \(z_{g}\) could come from differential expression analysis
Fast GSEA (FGSEA)

We’d like a representative score for genes within each set

Aggregating z-scores within a set
Function to run a t test for each gene:
Function to compute Stouffer Z-score


Constructing the null distribution by gene permutation
What if we don’t know the distribution of set-wise scores?

How it goes:
Repeatedly permuting gene set membership matrix while preserving the number of genes within each set
Compute set-level z-scores (or a similar kind) and construct null distribution
Calculate p-values by counting the frequency of observed \(S_{k}^{*} > S_{k}^{\textsf{perm}}\)
Constructing the null distribution by sample permutation

Repeat the permutation of case-control labels while preserving the same number of cases and controls
Compute set-level z-scores (or a similar kind) and construct null distribution
Calculate p-values by counting the frequency of observed \(S_{k}^{*} > S_{k}^{\textsf{perm}}\)
Summary of rank-based gene set analysis
When does it apply?
We have ranked all our genes by a score/test statistic (or compute a summary score for every gene set of interest)
We want to know whether any of our gene sets of interest tend to have more highly ranked genes (or higher summary scores)
When does it not apply?
- There is a hidden factor that can affect two steps: (1) gene set selection (annotations/knowledge) and (2) gene scoring
Note
Estimating the null distribution of ranks/scores over gene sets of interest is nontrivial